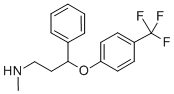
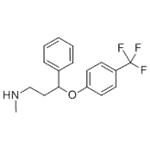
Fluoxetine
- Product NameFluoxetine
- CAS54910-89-3
- MFC17H18F3NO
- MW309.33
- EINECS611-209-7
- MOL File54910-89-3.mol
Chemical Properties
| Melting point | 158 °C |
| Boiling point | 395.1±42.0 °C(Predicted) |
| Density | 1.159±0.06 g/cm3(Predicted) |
| storage temp. | 2-8°C(protect from light) |
| solubility | 12.5mg/mL in DMSO, 16mg/mL in DMF, 12.5mg/mL in Ethanol |
| pka | 10.05±0.10(Predicted) |
| CAS DataBase Reference | 54910-89-3(CAS DataBase Reference) |
| NIST Chemistry Reference | Fluoxetine(54910-89-3) |
| EPA Substance Registry System | Benzenepropanamine, N-methyl-?-[4-(trifluoromethyl)phenoxy]- (54910-89-3) |
MSDS
| Provider | Language |
|---|---|
| (+/-)-N-Methyl-gamma-(4-(trifluoromethyl)phenoxy)benzenepropanamine | English |
Usage And Synthesis
Fluoxetine Hydrochloride: White or white crystalline solids with a melting point of 158.4 to 158.9℃. Soluble in methanol or ethanol, dissolved in acetonitrile, acetone, or chloroform, and slightly soluble in ethyl acetate, dichloromethane, or water(with sonication at pHl.2,4.5,7.0).Almost insoluble in cyclohexane, hexane or toluene.
Solubility(mg/m1):Methanol and ethanol > 100, Acetone, acetonitrile and chloroform 33~100, dichloromethane 5~10, water 1~2, ethyl acetate 2 ~ 2.5, Cyclohexane, hexane and toluene 0.5 ~ 0.67. The maximum solubility in water: 14mg/ml. UV maximum absorption (methanol):227,264,268,275nm (E1cm1%372.0,29.2,29.3,21.5).
Acute toxic of LD50 for large rate and small rat (mg/kg): 248, 452 oral administrations.

Solubility(mg/m1):Methanol and ethanol > 100, Acetone, acetonitrile and chloroform 33~100, dichloromethane 5~10, water 1~2, ethyl acetate 2 ~ 2.5, Cyclohexane, hexane and toluene 0.5 ~ 0.67. The maximum solubility in water: 14mg/ml. UV maximum absorption (methanol):227,264,268,275nm (E1cm1%372.0,29.2,29.3,21.5).
Acute toxic of LD50 for large rate and small rat (mg/kg): 248, 452 oral administrations.
Fluoxetine is an antidepressant of the selective serum reabsorption inhibitor (SSRI) type.The drug takes the form of Fluoxetine hydrochloride. Its trade name is "Prozac". The drug was developed by the Eli Lilly and Company. It has been launched into market for sale since the 1990s. This drug is used for the treatment of adult depression, obsessive-compulsive disorder, bulimia nervosa and the panic disorder that has or does not have the phobia of the square. It has good antidepressant effect and is widely used in clinical practice as a first-line antidepressant.The main pharmacological effect is to selectively inhibit the reuptake of 5 hydroxytryptamine (HT), which is released before the synapse of the central nervous system.It is also known as a selective 5- serotonin reuptake inhibitor. For other receptors, such as alpha adrenergic, beta adrenergic, 5- serotonin, and dopamine, fluoxetine had almost no binding force. Fluoxetine is well absorbed from the gastrointestinal tract after oral administration.The plasma concentration is about 6~8 hours, and eating does not affect its bioavailability. About 95% are combined with plasma proteins and are widely distributed.After taking the medicine for several weeks, the steady plasma concentration will be reached.By metabolism of the liver, the active metabolite, norfluoxetine is generated by demethylation. The half-life of fluoxetine is 4-6 days, and norfluoxetine is 4-16 days.It was mainly excreted by kidney. Because it can be secreted to breast milk, it is suggested to be cautious to pregnant women and breast-feeding women.
- There are a lot of side effects and the common adverse reactions are: systemic or local irritation, gastrointestinal disorders (such as nausea, vomiting, indigestion, diarrhea, dysphagia), anorexia, dizziness, headache, fatigue, sleep disorders, abnormal mental status, sexual dysfunction, abnormal vision, dyspnea etc..
- Fluoxetine should be banned for those who are using drugs such as monoamine oxidase inhibitor (MAOI).
- For hepatic insufficiency, the half-life of fluoxetine and norfluoxetine is increased to 7 days and 14 days respectively. Therefore, it is important to consider reducing the dosage or reducing the frequency of drug use.
Prozac was discovered by a team of chemists at the pharmaceutical company Eli Lilly. Key
researchers involved in the work were Bryan B. Molloy (1939–2004), Klaus K. Schmiegel
(1939–), Ray W. Fuller (1935–1996), and David T. Wong (1935–). In the middle of the
20th century, the main group of drugs for treating depression was tricyclic antidepressants
(TCAs) and monoamine oxidase inhibitors (MAOIs). TCAs are named because of their
three-ring chemical structure. Lilly researchers were working with TCAs in the1950s and
1960s. Prozac was developed by Eli Lilly scientists who based their work on the antihistamine diphenylhydramine; diphenylhydramine hydrochloride is marketed under the trade name
Benadryl.the Lilly scientists examined diphenylhydramine because research had demonstrated
that some antihistamines, including diphenylhydramine, had the ability to inhibit
serotonin and serve as antidepressants.Molloy started examining diphenylhydramine-type
compounds for their antidepressant properties in 1970. Molloy and his colleagues discovered
fluoxetine hydrochloride had potential as an antidepressant in 1972 and it was referred to
as Lilly 110140 in thefirst published articles on the compound, which appeared in 1974.
Fluoxetine hydrochloride was no more effective than other antidepressant drugs of the time,
but it produced much fewer negative side effects because it interacted specifically with the neurotransmitter
serotonin but did not interfere with other neurotransmitters. TCAs inhibited
the reuptake of other neurotransmitters along with serotonin. Molloy and Schmiegel applied
for a patent in 1974 for the synthesis of arloxyphenylpropylamines (U.S. Patent Number
4314081).the patent named a number of compounds in this class of chemicals that could
be used as antidepressants. In 1983, Eli Lilly applied to the Food and Drug Administration
(FDA) for approval of fluoxetine hydrochloride as a drug used to treat depression. Prozac was
first offered to the public in Belgium in 1986 and in the United States in 1988. Eli Lilly initially
had a monopoly on fluoxetine hydrochloride as an antidepressant with its Prozac brand.
In the mid-1990s, a lawsuit filed against Eli Lilly led to the loss of their exclusive patent rights,
allowing generic fluoxetine hydrochloride antidepressants to be marketed starting in 2001.
Fluoxetine in its hydrochloride salt form is marketed as numerous drugs, the most popular ofwhich is Prozac. Prozac is prescribed for depression, obsessive-compulsive disorder, bulimia,agoraphobia, and premenstrual dysphoric disorder (premenstrual syndrome). Prozac andother fluoxetine medications belong to a class of drugs called selective serotonin reuptakeinhibitors (SSRIs). When a nerve signal is sent, a neurotransmitter, such as serotonin, travelsfrom a presynaptic neuron across the synaptic gap to a postsynaptic neuron. Receptors on thepostsynaptic neuron capture the neurotransmitter, resulting in the transmission of the signal.After performing its function, the neurotransmitter is released back to the presynaptic cellin a process called reuptake. SSRIs slow down the return of serotonin to presynaptic neurons,allowing for a higher serotonin concentration on postsynaptic neurons. Because depressionand other psychological disorders are associated with low serotonin levels, Prozac and otherSSRIs help maintain serotonin levels.
Prozac was thefirst SSRI antidepressant to be marketed. Because Prozac produced lesssevere side effects than other antidepressants, it became the drug of choice for treating depressionand was made available to a wider public. Its use exploded in the 1990s, with sales peakingin 2000 when revenues from Prozac reached $2.5 billion. Eli Lilly’s patent on fluoxetinehydrochloride expired in August 2001; its use continued into the 21st century but on a muchsmaller scale as generic fluoxetine hydrochloride products came on the market. Since its introductionin 1986, Prozac was the most prescribed drug for antidepressant until recent yearswhen it was replaced by Zoloft, Paxil, and Lexapro as the top three antidepressants prescribedin the United States, respectively.
Fluoxetine hydrochloride is most recognized as an antidepressant, but it is also used torelieve symptoms of premenstrual dysphoric disorder (PMDD) (premenstrual syndrome).Th ese symptoms include mood swings, tension, bloating, irritability, and breast tenderness. EliLilly began marketing fl uoxetine hydrochloride as Sarafem in 2000 for treating PMDD.
Prozac was thefirst SSRI antidepressant to be marketed. Because Prozac produced lesssevere side effects than other antidepressants, it became the drug of choice for treating depressionand was made available to a wider public. Its use exploded in the 1990s, with sales peakingin 2000 when revenues from Prozac reached $2.5 billion. Eli Lilly’s patent on fluoxetinehydrochloride expired in August 2001; its use continued into the 21st century but on a muchsmaller scale as generic fluoxetine hydrochloride products came on the market. Since its introductionin 1986, Prozac was the most prescribed drug for antidepressant until recent yearswhen it was replaced by Zoloft, Paxil, and Lexapro as the top three antidepressants prescribedin the United States, respectively.
Fluoxetine hydrochloride is most recognized as an antidepressant, but it is also used torelieve symptoms of premenstrual dysphoric disorder (PMDD) (premenstrual syndrome).Th ese symptoms include mood swings, tension, bloating, irritability, and breast tenderness. EliLilly began marketing fl uoxetine hydrochloride as Sarafem in 2000 for treating PMDD.
ChEBI: N-methyl-3-phenyl-3-[4-(trifluoromethyl)phenoxy]propan-1-amine is an aromatic ether consisting of 4-trifluoromethylphenol in which the hydrogen of the phenolic hydroxy group is replaced by a 3-(methylamino)-1-phenylpropyl group. It is a member of (trifluoromethyl)benzenes, an aromatic ether and a secondary amino compound.
About 600 g of β-dimethylaminopropiophenone hydrochloride were converted to the corresponding free base by the action of 1.5 N aqueous sodium hydroxide. The liberated free base was taken up in ether, the ether layer separated and dried, and the ether removed therefrom in vacuo. The residual oil comprising β-dimethylaminopropiophenone was dissolved in 2 L of tetrahydrofuran, and the resulting solution added in dropwise fashion with stirring to a solution of four moles of diborane in 4 L of tetrahydrofuran. The reaction mixture was stirred overnight at room temperature. An additional mole of diborane in 1 L of tetrahydrofuran was added, and the reaction mixture stirred again overnight at room temperature. Next, 2 L of aqueous hydrochloric acid were added to decompose any excess diborane present. The tetrahydrofuran was removed by evaporation. The acidic solution was extracted twice with 1 L portions of benzene, and the benzene extracts were discarded. The acidic solution was then made basic with an excess of 5 N aqueous sodium hydroxide. The basic solution was extracted three times with 2 L portions of benzene. The benzene extracts were separated and combined, and the combined extracts washed with a saturated aqueous sodium chloride and then dried. Evaporation of the solvent in vacuo yields 442 g of N,Ndimethyl-3-phenyl-3-hydroxypropylamine.
A solution containing 442 g of N,N-dimethyl-3-phenyl-3-hydroxypropylamine in 5 L of chloroform was saturated with dry gaseous hydrogen chloride. 400 ml of thionyl chloride were then added to the chloroform solution at a rate sufficient to maintain reflux. The solution was refluxed an additional 5 h. Evaporation of the chloroform and other volatile constituents in vacuo yielded N,N-dimethyl-3-phenyl-3-chloropropylamine hydrochloride which was collected by filtration, and the filter cake washed twice with 1500 ml portions of acetone. The washed crystals weighed about 500 g and melted at 181°-183°C with decomposition. An additional 30 g of compound were obtained from the acetone wash by standard crystallization procedures. The structure of the above compound was verified by NMR and titration.
A solution of 50 g p-trifluoromethylphenol, 12 g of solid sodium hydroxide and 400 ml of methanol was placed in a 1 L round-bottom flask equipped with magnetic stirrer, condenser and drying tube. The reaction mixture was stirred until the sodium hydroxide had dissolved. Next, 29.8 g of N,N-dimethyl-3phenyl-3-chloropropylamine hydrochloride were added. The resulting reaction mixture was refluxed for about 5 days and then cooled. The methanol was then removed by evaporation, and the resulting residue taken up in a mixture of ether and 5 N aqueous sodium hydroxide. The ether layer was separated and washed twice with 5 N aqueous sodium hydroxide and three times with water. The ether layer was dried, and the ether removed by evaporation invacuo to yield as a residue N,N-dimethyl-3-(p-trifluoromethylphenoxy)-3phenylpropylamine.
A solution containing 8.1 g of cyanogen bromide in 500 ml benzene and 50 ml of toluene was placed in a 1 L three-neck round-bottom flask equipped with thermometer, addition funnel, drying tube and inlet tube for nitrogen. The solution was cooled to about 5°C with stirring, and nitrogen gas was bubbled thru the solution. Next, a solution of 12.146 g of N,N-dimethyl-3-(ptrifluoromethylphenoxy)-3-phenylpropylamine dissolved in 40 ml of benzene was added in dropwise fashion. The temperature of the reaction mixture was allowed to rise slowly to room temperature, at which temperature stirring was continued overnight while still maintaining a nitrogen atmosphere 100 ml of benzene were added. The reaction mixture was washed twice with water, once with 2 N aqueous sulfuric acid and then with water until neutral. The organic layer was dried, and the solvents removed therefrom by evaporation in vacuo to yield about 9.5 g of an oil comprising N-methyl-N-cyano-3-(ptrifluoromethylphenoxy)-3-phenylpropylamine.
A solution of 100 g potassium hydroxide, 85 ml water, 400 ml ethylene glycol and 9.50 g of N-methyl-N-cyano-3-(p-trifluoromethylphenoxy)-3phenylpropylamine was prepared in a 1 L three-neck, round-bottom flask equipped with magnetic stirrer and condenser. The reaction mixture was heated to refluxing temperature (130°C) for 20 h, and was then cooled. 500 ml of water were added. The reaction mixture was extracted with three 500 ml portions of ether. The ether extracts were combined, and the combined extracts washed with water. The water wash was discarded. The ether solution was next contacted with 2 N aqueous hydrochloric acid. The acidic aqueous layer was separated. A second aqueous acidic extract with 2 N hydrochloric acid was made followed by three aqueous extracts and an extract with saturated aqueous sodium chloride. The aqueous layers were all combined and made basic with 5 N aqueous sodium hydroxide. N-Methyl-3-(ptrifluoromethylphenoxy)-3-phenylpropylamine, formed in the above reaction, was insoluble in the basic solution and separated. The amine was extracted into ether. Two further ether extractions were carried out. The ether extracts were combined, and the combined extracts washed with saturated aqueous sodium chloride and then dried. Evaporation of the ether in vacuo yielded about 6.3 g of N-methyl-3-(p-trifluoromethylphenoxy)-3-phenylpropylamine.
A solution containing 442 g of N,N-dimethyl-3-phenyl-3-hydroxypropylamine in 5 L of chloroform was saturated with dry gaseous hydrogen chloride. 400 ml of thionyl chloride were then added to the chloroform solution at a rate sufficient to maintain reflux. The solution was refluxed an additional 5 h. Evaporation of the chloroform and other volatile constituents in vacuo yielded N,N-dimethyl-3-phenyl-3-chloropropylamine hydrochloride which was collected by filtration, and the filter cake washed twice with 1500 ml portions of acetone. The washed crystals weighed about 500 g and melted at 181°-183°C with decomposition. An additional 30 g of compound were obtained from the acetone wash by standard crystallization procedures. The structure of the above compound was verified by NMR and titration.
A solution of 50 g p-trifluoromethylphenol, 12 g of solid sodium hydroxide and 400 ml of methanol was placed in a 1 L round-bottom flask equipped with magnetic stirrer, condenser and drying tube. The reaction mixture was stirred until the sodium hydroxide had dissolved. Next, 29.8 g of N,N-dimethyl-3phenyl-3-chloropropylamine hydrochloride were added. The resulting reaction mixture was refluxed for about 5 days and then cooled. The methanol was then removed by evaporation, and the resulting residue taken up in a mixture of ether and 5 N aqueous sodium hydroxide. The ether layer was separated and washed twice with 5 N aqueous sodium hydroxide and three times with water. The ether layer was dried, and the ether removed by evaporation invacuo to yield as a residue N,N-dimethyl-3-(p-trifluoromethylphenoxy)-3phenylpropylamine.
A solution containing 8.1 g of cyanogen bromide in 500 ml benzene and 50 ml of toluene was placed in a 1 L three-neck round-bottom flask equipped with thermometer, addition funnel, drying tube and inlet tube for nitrogen. The solution was cooled to about 5°C with stirring, and nitrogen gas was bubbled thru the solution. Next, a solution of 12.146 g of N,N-dimethyl-3-(ptrifluoromethylphenoxy)-3-phenylpropylamine dissolved in 40 ml of benzene was added in dropwise fashion. The temperature of the reaction mixture was allowed to rise slowly to room temperature, at which temperature stirring was continued overnight while still maintaining a nitrogen atmosphere 100 ml of benzene were added. The reaction mixture was washed twice with water, once with 2 N aqueous sulfuric acid and then with water until neutral. The organic layer was dried, and the solvents removed therefrom by evaporation in vacuo to yield about 9.5 g of an oil comprising N-methyl-N-cyano-3-(ptrifluoromethylphenoxy)-3-phenylpropylamine.
A solution of 100 g potassium hydroxide, 85 ml water, 400 ml ethylene glycol and 9.50 g of N-methyl-N-cyano-3-(p-trifluoromethylphenoxy)-3phenylpropylamine was prepared in a 1 L three-neck, round-bottom flask equipped with magnetic stirrer and condenser. The reaction mixture was heated to refluxing temperature (130°C) for 20 h, and was then cooled. 500 ml of water were added. The reaction mixture was extracted with three 500 ml portions of ether. The ether extracts were combined, and the combined extracts washed with water. The water wash was discarded. The ether solution was next contacted with 2 N aqueous hydrochloric acid. The acidic aqueous layer was separated. A second aqueous acidic extract with 2 N hydrochloric acid was made followed by three aqueous extracts and an extract with saturated aqueous sodium chloride. The aqueous layers were all combined and made basic with 5 N aqueous sodium hydroxide. N-Methyl-3-(ptrifluoromethylphenoxy)-3-phenylpropylamine, formed in the above reaction, was insoluble in the basic solution and separated. The amine was extracted into ether. Two further ether extractions were carried out. The ether extracts were combined, and the combined extracts washed with saturated aqueous sodium chloride and then dried. Evaporation of the ether in vacuo yielded about 6.3 g of N-methyl-3-(p-trifluoromethylphenoxy)-3-phenylpropylamine.
Fluoxetine (Prozac) is given in the morning because of
its potential for being activating and causing insomnia.
Food does not affect its systemic bioavailability and
may actually lessen the nausea reported by some patients.
Fluoxetine is highly bound to serum proteins and
may interact with other highly protein bound drugs. It is
demethylated in the liver to form an active metabolite,
norfluoxetine. Inactive metabolites are excreted by the
kidney.Doses must be reduced in patients with liver disease.
The slow elimination of fluoxetine and norfluoxetine lead to special clinical concerns when adjusting doses and discontinuing this medication. Steady state is not reached until 4 to 6 weeks, and similarly, complete elimination takes 4 to 6 weeks after discontinuation of the medication. A 4- to 6-week waiting period should be permitted before starting a medication with potential for an interaction with fluoxetine, such as a monoamine oxidase inhibitor (MAOI). Additionally, fluoxetine is a potent inhibitor of cytochrome P450 2D6 and can significantly elevate levels of drugs metabolized by this route. Thus, coadministration of drugs with a narrow therapeutic index, such as TCAs and type 1C antiarrhythmics, including flecainide and propafenone, are a particular concern.
The slow elimination of fluoxetine and norfluoxetine lead to special clinical concerns when adjusting doses and discontinuing this medication. Steady state is not reached until 4 to 6 weeks, and similarly, complete elimination takes 4 to 6 weeks after discontinuation of the medication. A 4- to 6-week waiting period should be permitted before starting a medication with potential for an interaction with fluoxetine, such as a monoamine oxidase inhibitor (MAOI). Additionally, fluoxetine is a potent inhibitor of cytochrome P450 2D6 and can significantly elevate levels of drugs metabolized by this route. Thus, coadministration of drugs with a narrow therapeutic index, such as TCAs and type 1C antiarrhythmics, including flecainide and propafenone, are a particular concern.
In fluoxetine (Prozac), protonated in vivo, the protonatedamino group can H-bond to the ether oxygen electrons, whichcan generate the β-arylamino–like group, with the other arylserving as the characteristic “extra” aryl. The S-isomer ismuch more selective for SERT than for NET. The majormetabolite is the N-demethyl compound, which is as potent asthe parent and more selective (SERT versus NET).
Therapy for 2 or more weeks is required for the antidepressanteffect. Somatodendritic 5-HT1A autoreceptor desensitizationwith chronic exposure to high levels of 5-HT isthe accepted explanation for the delayed effect for this andother serotonin reuptake inhibitors.
Therapy for 2 or more weeks is required for the antidepressanteffect. Somatodendritic 5-HT1A autoreceptor desensitizationwith chronic exposure to high levels of 5-HT isthe accepted explanation for the delayed effect for this andother serotonin reuptake inhibitors.
Fluoxetine is a potent and selective inhibitor of 5-HT reuptake, but not of NE or dopamine uptake in the CNS.
Its mechanism of action is common to the SSRIs. Fluoxetine does not interact directly with postsynaptic 5-HT
receptors and has weak affinity for the other neuroreceptors. Both enantiomers of fluoxetine display similar
affinities for human SERT. The NE:5-HT selectivity ratio, however, indicates that the S-enantiomer is
approximately 100 times more selective for SERT inhibition than the R-enantiomer. The R-(+)-stereoisomer is
approximately eight times more potent an inhibitor of SERT together with a longer duration of action than the
S-(–)-isomer. However, the S-(–)-norfluoxetine metabolite is seven times more potent as an inhibitor of the
5-HT transporter than the R-(+)-metabolite, with a selectivity ratio approximately equivalent to that of
S-fluoxetine.
The pharmacokinetics of fluoxetine fit the general characteristics of the SSRIs. Of particular
importance is its long half-life contributing to its nonlinear pharmacokinetics. In vitro studies show that
fluoxetine and norfluoxetine are potent inhibitors of CYP2D6 and CYP3A4 and less potent inhibitors of
CYP2C9, CYP2C19 and CYP1A2. Fluoxetine is metabolized primarily by CYP2D6 N-demethylation to its active
metabolite norfluoxetine and, to a lesser extent, O-dealkylation to form the inactive metabolite
p-trifluoromethylphenol. Following oral administration, fluoxetine and its metabolites are excreted principally
in urine, with approximately 73% as unidentified metabolites, 10% as norfluoxetine, 10% as norfluoxetine
glucuronide, 5% as fluoxetine N-glucuronide, and 2% as unmetabolized drug.
Both R- and S-Norfluoxetine were less potent than the corresponding enantiomers of fluoxetine as inhibitors of NE uptake. Inhibition of 5-HT uptake in cerebral cortex persisted for more than 24 hours after administration of S-norfluoxetine similarly to fluoxetine. Thus, S-norfluoxetine is the active N-demethylated metabolite responsible for the persistently potent and selective inhibition of 5-HT uptake in vivo.
The pharmacokinetics of fluoxetine in healthy geriatric individuals do not differ substantially from those in younger adults. Because of its relatively long half-life and nonlinear pharmacokinetics, the possibility of altered pharmacokinetics in geriatric individuals could exist, particularly those with systemic disease and/or in those receiving multiple medications concurrently. The elimination half-lives of fluoxetine and norfluoxetine do not appear to be altered substantially in patients with renal or hepatic impairment.
Both R- and S-Norfluoxetine were less potent than the corresponding enantiomers of fluoxetine as inhibitors of NE uptake. Inhibition of 5-HT uptake in cerebral cortex persisted for more than 24 hours after administration of S-norfluoxetine similarly to fluoxetine. Thus, S-norfluoxetine is the active N-demethylated metabolite responsible for the persistently potent and selective inhibition of 5-HT uptake in vivo.
The pharmacokinetics of fluoxetine in healthy geriatric individuals do not differ substantially from those in younger adults. Because of its relatively long half-life and nonlinear pharmacokinetics, the possibility of altered pharmacokinetics in geriatric individuals could exist, particularly those with systemic disease and/or in those receiving multiple medications concurrently. The elimination half-lives of fluoxetine and norfluoxetine do not appear to be altered substantially in patients with renal or hepatic impairment.
Fluoxetine is a phenylpropylamine that inhibits the neuronal reuptake of serotonin, which
presumably has a direct relationship on antidepressant activity. This compound has either no effect or a small effect on the neuronal reuptake of norepinephrine and dopamine. In
addition, it does not bind to cholinergic, histaminergic, or α-adrenergic receptors, which is
believed to be the cause of tricyclic antidepressant side effects.
Fluoxetine is a 3-phenoxy-3-phenylpropylamine that exhibits selectivity and high affinity for human SERT and
low affinity for NET. It is marketed as a racemic mixture of R- and S-fluoxetine. Its selectivity for
SERT inhibition depends on the position of the substituent in the phenoxy ring.
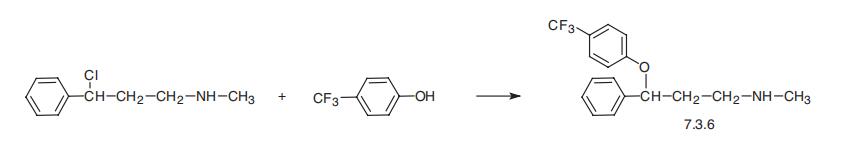
Fluoxetine, 3-[p-(trifluoromethyl)-phenoxy]-N-methyl-3-phenylpropylamine
(7.3.6), is synthesized by reaction of p-trifluoromethylphenol with 3-(chloro)-N-methyl-3-
phenylpropylamine in the presence of potassium carbonate [59,60].
Fluoxetine and its norfluoxetine metabolite, like many other drugs metabolized by CYP2D6, inhibit the activity
of CYP2D6 and, potentially, may increase plasma concentrations of concurrently administered drugs that also
are metabolized by this enzyme. Fluoxetine may make normal CYP2D6 metabolizers resemble poor
metabolizers. Fluoxetine can inhibit its own CYP2D6 metabolism, resulting in higher-than-expected plasma
concentrations during upward dose adjustments. Therefore, switching from fluoxetine to another SSRI or
other serotonergic antidepressant requires a washout period of at least 5 weeks or a lowerthan-recommended initial dose with monitoring for adverse events.
Fluoxetine is highly protein bound and may affect the free plasma concentration and, thus, the pharmacological effect of other highly protein-bound drugs (e.g., warfarin sodium).
Fluoxetine is highly protein bound and may affect the free plasma concentration and, thus, the pharmacological effect of other highly protein-bound drugs (e.g., warfarin sodium).
Fluoxetine is extensively metabolised by the enzyme
CYP2D6 in the liver to its primary active metabolite
norfluoxetine (desmethylfluoxetine), by desmethylation.
The elimination half-life of fluoxetine is 4-6 days and for
norfluoxetine 4-6 days. Excretion is mainly (about 60%)
via the kidney.
Preparation Products And Raw materials
Raw materials
- Thionyl chlorideMethylaminePotassium iodideParaformaldehydeAcetophenonePotassium borohydrideN,N-DimethylacetamideBenzotrifluoridePropiophenone4-ChlorobenzotrifluorideCyanogen bromideDiborane4-FluorophenolFluoxetine hydrochloride3-Hydroxy-N-methyl-3-phenyl-propylamineSulfuric acid Sodium hydroxideEthylene glycolHydrochloric acid4-TrifluoromethylphenolPotassium hydroxide
Fluoxetine manufacturers


Related Product Information
PROMPT×
PROMPT
The What'sApp is temporarily not supported in mainland China
The What'sApp is temporarily not supported in mainland China
Cancel
Determine