A brief introduction to Cefiderocol
Description
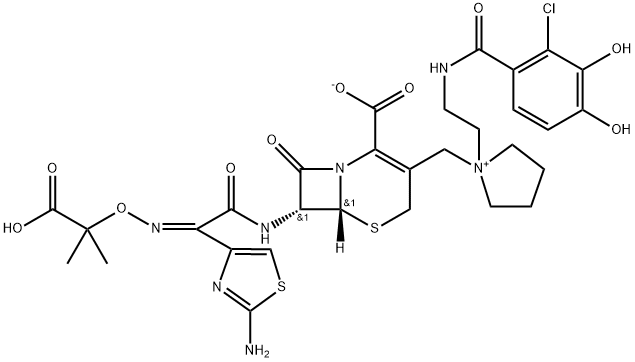
Cefiderocol, formerly S-649266, is a novel catechol-substituted siderophore cephalosporin for injection that uses the bacterial active iron transport channels to penetrate the outer membrane and enter the periplasmic space. Cefiderocol is structurally different from other siderophore-conjugated monobactam antibiotics, such as BAL30072 and MC-1, which are conjugated to a dihydroxypyridone siderophore[1-2].
Biological activity
The potent activity of cefiderocol is partly a result of its high stability against various ESBLs and serine-type and metallo-type carbapenemases. Like other β-lactam antibiotics, cefiderocol consists of a 4-membered β-lactam ring bound to a 6-membered dihydrothiazine ring. The 3-position side chain in cefiderocol consists of a catechol 2-chloro3,4-dihydroxybenzoic acid moiety that is covalently bound via a linker to the pyrrolidine ring, which is identical to the pyrrolidinium group found in cefepime. This quaternized N-methyl-pyrrolidine moiety at the 3′ position confers zwitterionic properties that enhance water solubility and allow the molecule to rapidly penetrate the outer cell membrane of Gram-negative bacteria. In addition, the moiety has no inducing capacity and very low affinity to β-lactamases, making it less recognizable and resistant to degradation.
Synthetic method
Although syntheses of cefiderocol and its analogs that are suitable for medicinal chemistry purposes have been reported, a convergent approach involving the linkage of a catechol-containing fragment linked through a quaternary pyrrolidinium salt to a β-lactam on a multikilogram scale was reported by Shionogi & CO [3].
Synthesis of Cefiderocol Protected Catechol Fragment 7

Preparation of the catechol fragment 7 began with chlorination of benzaldehyde 1. Demethylation of 2 with AlCl3 afforded catechol 3, which was then protected as p-methoxybenzyl (PMB) ether 4 prior to oxidation to give benzoic acid 5. Finally, activating the acid with mesyl chloride prior to coupling with amine 6 gave the protected fragment 7 in 62% overall yield over five steps from 1.
Synthesis of Cefiderocol

The construction of the β-lactam subunit and its linkage to catechol fragment 7 began with aminothiazole 8, whose substructure is also incorporated into other β-lactam antibiotics such as ceftazidime (Fortaz) and aztreonam (Azactam). The acid was activated with mesyl chloride before reaction with the cephalosporin core 9, obtained semisynthetically from a fermentation precursor. Sulfide 10 was then carefully oxidized to sulfoxide 11 to minimize competing reactions during the subsequent quaternization step. Exposure of chloride 11 to pyrrolidine 7 in the presence of boric acid and sodium iodide afforded 12 as the tetraalkylammonium iodide salt. The sulfoxide was then converted back to the sulfide 13 upon treatment with phosphorus trichloride. Acidic removal of the PMB groups on a multikilogram scale followed by crystallization with sulfuric acid and p-toluenesulfonic acid gave cefidicerol. Although the yields and conditions outlined in Schemes 1 and 2 reflect those exemplified in the patent, the authors noted that alternative reagents can perform the same transformations for this sequence.
References
[1] Rania M El-Lababidi, John George Rizk. “Cefiderocol: A Siderophore Cephalosporin.” Annals of Pharmacotherapy 54 12 (2020): 1215–1231.
[2] Christopher Fröhlich. “Evolution of β-lactamase-mediated cefiderocol resistance.” Journal of Antimicrobial Chemotherapy (2022): 2429–2436.
[3] Andrew C. Flick. “Synthetic Approaches to the New Drugs Approved during 2019.” Journal of Medicinal Chemistry 64 7 (2021): 3604–3657.
);