How to synthesize Eravacycline?
Descriotion
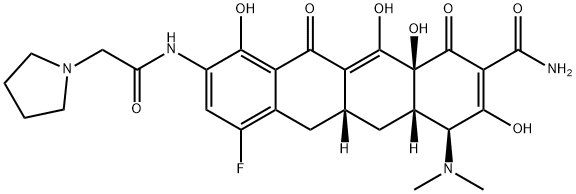
Eravacycline belongs to the tetracycline class of antibiotics and was approved by the United States Food and Drug Administration (USFDA) for treating complicated intra-abdominal infections in patients 18 years and older. Eravacycline is a fully synthetic broad-spectrum antibiotic that exhibits potent activity against Gram-positive and Gram-negative bacterial strains, including many that have acquired tetracycline-specific resistant mechanisms.
Synthetic method
Professor Andrew Myers' laboratory at Harvard University described a highly convergent route for preparing this compound. Retrosynthetically, eravacycline was envisioned to be derived via a Michael addition−Dieckmann cyclization reaction between the anion of compound 1 and Michael acceptor 2 (as shown below)[1]. Tricyclic intermediate 2 was envisioned to come from adding the anion of isoxazole 4 to the aldehyde 3, followed by an intramolecular Diels−Alder reaction to set the carbon framework 2. Isoxazole 4 was envisioned to come from dimethyl maleate, and the chiral vinyl amine stereocenter was set via an Ellman sulfinamide auxiliary.

Preparation of Eravacycline Isoxazole 4 (4· Tartrate)

The preparation of the chiral isoxazole 4 is described in Scheme 1. Dimethyl maleate (5) was treated with bromine in the presence of azo-bis(isobutyronitrile) (AIBN) and ultraviolet light to give dibromide 6. Condensation of 6 with hydroxyurea in the presence of potassium tert-butoxide provided isoxazole 7. Benzylation of the hydroxy group of 7 followed by DIBAL reduction of the ester gave aldehyde 8 in high yield over the two steps. Condensation of 8 with (S)-tert-butylsulfinylamide (Ellman’s auxiliary) in the presence of copper(II) sulfate provided chiral sulfinimine 9. After reaction optimization, 9 was treated with vinylmagnesium chloride in the presence of methyllithium and zinc chloride to give 10. The tertbutylsulfinyl group was removed under acidic conditions. The resulting primary amine was treated with formaldehyde in the presence of sodium acetate and then reduced using a picoline-borane complex to give the dimethylamine coupling partner 4. The ee was enhanced to 99.0% by tartrate salt formation, giving 4· Tartrate.
Construction of Eravacycline Tricyclic Michael Acceptor Enone 2
Treating 4· Tartrate with sodium hydroxide provided the free base 4, which was reacted with tetramethylpiperidine (TMP) magnesium chloride−lithium chloride complex to effect the direct magnesiation of 4 on the oxazole ring, with no competing allylic metalation. This intermediate was reacted with aldehyde 3 to give alcohols 11a/ 11b. Heating the mixture of 11a/11b in DMSO, DIPEA, butylated hydroxytoluene, and isopropyl acetate affected the intramolecular Diels−Alder reaction to give a mixture of endo products (12a/12b), arising from 11a, and a mixture of Exo products (12c/12d), arising from 11b. This mixture of alcohols 12a−d was oxidized with sulfur trioxide pyridine complex to give ketones 13a/13b in 99.1:0.9 dr. Treating 13a/13b with boron trichloride efficiently affected the demethylation of the methyl enol ether, which spontaneously underwent ring opening to provide enone 14 in high yield. Protection of the resulting alcohol as its tributylsilyl ether followed by recrystallization from isopropyl alcohol provided the tricyclic Michael acceptor coupling partner 2.

Preparation of Dibenzyl Amine Protected Coupling Partner 1

Compound 15 was treated initially with LDA and then quenched with methyl iodide to give arene 16. Formation of the acid chloride of 16 followed by reaction with phenol provided the corresponding ester, making way for methyl ether cleavage with boron tribromide to provide phenol 17. Nitration of 17 and protection of the phenol as a benzyl ether gave nitroarene 18. Reduction of the nitro group with sodium bisulfite provided aniline 19 from compound 15. Amine 19 reacted with benzyl bromide to produce the dibenzylamine-protected coupling partner 1 in an 80% yield.
Final Assembly of Eravacycline
The completion of the synthesis of eravacycline is described below. Compound 1 was treated with LDA, and compound 2 was to promote the desired intermolecular Michael addition. The resulting Michael adduct was then treated with lithium bis(trimethylsilyl)amide (LHMDS) to induce an intramolecular Dieckmann cyclization, which gave compound 20. The silyl-protecting group was removed using hydrofluoric acid, and the resulting intermediate was treated with hydrogen and palladium on carbon. These conditions resulted in removing the dibenzylamine and benzyl ether protecting groups, which further gave rise to the opening of the oxazole ring to arrive at compound 21 for the two-step sequence. Compound 21 was then reacted with acid chloride 22 to furnish eravacycline in 89% yield.

References
[1] Andrew C. Flick. “Synthetic Approaches to New Drugs Approved during 2018.” Journal of Medicinal Chemistry 63 19 (2020): 10652–10704.
);
You may like
See also


US $0.00-0.00/kg2023-12-11
- CAS:
- 1207283-85-9
- Min. Order:
- 1kg
- Purity:
- 99%
- Supply Ability:
- 100tons