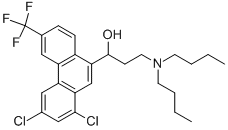
HALOFANTRINE
- Product NameHALOFANTRINE
- CAS69756-53-2
- MFC26H30Cl2F3NO
- MW500.42
- EINECS274-104-1
- MOL File69756-53-2.mol
Chemical Properties
| Density | 1.244±0.06 g/cm3 (20 ºC 760 Torr) |
| storage temp. | 2-8°C |
| Water Solubility | 0.59mg/L(37 ºC) |
Usage And Synthesis
Halofantrine is a phenanthrenemethanol antimalarial drug developed by the US military. The antimalarial activity of the phenanthrenemethanols was discovered during the Second World War but was not exploited until later. Halofantrine is a potent blood schizontocide against Plasmodium falciparum both in vitro and in vivo. However, it has no effect against exoerythrocytic forms of the parasite. Experience of its activity against other malaria species is limited. The drug exists as a racemic mixture, but the two enantiomers have shown similar activity in vitro [1, 2].
The mechanism of action of halofantrine is not known.
The mechanism of action of halofantrine is not known.
Halofantrine is indicated only for the treatment of multidrug-resistant Plasmodium falciparum malaria.
Halofantrine is generally well tolerated. Mild and transient side effects such as nausea, vomiting, diarrhoea, abdominal pain, pruritus and rash have been reported in humans. Halofantrine is potentially cardiotoxic particularly with doses above the recommended dose and causes ECG changes such as prolongation of PR and QTc intervals. In one study, the sudden death of a patient was reported after receiving a high dose of halofantrine (8 mg/kg 3 times daily for 3 days) [3]. The patient had previously been treated with mefloquine. Cardiotoxicity due to halofantrine will become a therapeutic problem if higher dosage regimens have to be used due to decreased efficacy [3]. Occasional elevation of serum transaminase have been observed in some patients. The relationship of this to the treatment is unclear. Values usually return to normal levels within a week after treatment [4-6].
Halofantrine should not be given to patients with pre-existing cardiovascular diseases. There is a warning against the concomitant intake of any cardiotoxic drugs. Halofantrine is not used for malaria prophylaxis.
Available as halofantrine hydrochloride: 100 mg hydrochloride is equal to 93 mg base. Not yet available for parenteral use.
• Halfan® (SmithKline & Beecham). Tablets 250 mg. Oral suspension 20 mg/ml.
• Halfan® (SmithKline & Beecham). Tablets 250 mg. Oral suspension 20 mg/ml.
1. Bryson HM, Goa KL (1992). Halofantrine. A review of its antimalarial activity, pharmacokinetic properties and therapeutic potential. Drugs, 43, 236–258.
2. Karle JM, Olmeda R, Gerena L, Milhous WK (1994). Plasmodium falciparum: Role of absolute stereochemistry in the antimalarial activity of synthetic aminoalcohol antimalarial agents. Exp Parasitol, 76, 345–351.
3. Nosten F, ter Kuile FO, Luxemburger C, Woodrow C, Kyle DPE, Chongsuphajaisiddhi T, White NJ (1993). Cardiac effects of antimalarial treatment with halofantrine. Lancet, 341, 1054–1056.
4. Watkins WM, Lury JD, Kariuki D, Koech DK, Oloo JA, Mosoba M, Mjomba M, Gilles HM (1988). Efficacy of multiple-dose halofantrine in treatment of chloroquine-resistant falciparum malaria in children in Kenya. Lancet, 2, 247–250.
5. Salako LA, Sowunmi A, Walker O (1990). Evaluation of the Clinical trials and safety of halofantrine in falciparum malaria in Ibadan Nigeria. Trans R Soc Trop Med Hyg, 84, 644–647.
6. Boudreau EF, Pang LW, Dixon KE, Webster HK, Pavanand K, Tosingha L, Somutsakorn P, Canfield CJ (1988). Malaria: Treatment efficacy of halofantrine (WR171,669) in initial field trials in
7. Karbwang J, Na Bangchang K (1994). Clinical pharmacokinetics of halofantrine. Clin Pharmacokinet, 27(2), 104–119.
2. Karle JM, Olmeda R, Gerena L, Milhous WK (1994). Plasmodium falciparum: Role of absolute stereochemistry in the antimalarial activity of synthetic aminoalcohol antimalarial agents. Exp Parasitol, 76, 345–351.
3. Nosten F, ter Kuile FO, Luxemburger C, Woodrow C, Kyle DPE, Chongsuphajaisiddhi T, White NJ (1993). Cardiac effects of antimalarial treatment with halofantrine. Lancet, 341, 1054–1056.
4. Watkins WM, Lury JD, Kariuki D, Koech DK, Oloo JA, Mosoba M, Mjomba M, Gilles HM (1988). Efficacy of multiple-dose halofantrine in treatment of chloroquine-resistant falciparum malaria in children in Kenya. Lancet, 2, 247–250.
5. Salako LA, Sowunmi A, Walker O (1990). Evaluation of the Clinical trials and safety of halofantrine in falciparum malaria in Ibadan Nigeria. Trans R Soc Trop Med Hyg, 84, 644–647.
6. Boudreau EF, Pang LW, Dixon KE, Webster HK, Pavanand K, Tosingha L, Somutsakorn P, Canfield CJ (1988). Malaria: Treatment efficacy of halofantrine (WR171,669) in initial field trials in
7. Karbwang J, Na Bangchang K (1994). Clinical pharmacokinetics of halofantrine. Clin Pharmacokinet, 27(2), 104–119.
Halofantrine is an orally-active blood schizonticide reportedly highly effective in the
treatment of fulcipurum malaria and other types of parasitemia. Cure rate is claimed to be
over 95%.
ChEBI: 3-(dibutylamino)-1-[1,3-dichloro-6-(trifluoromethyl)-9-phenanthrenyl]-1-propanol is a member of phenanthrenes.
Halofantrine is an antimalarial introduced to medicine in 1982. It
should be reserved for use in areas where multiple drug-resistant falciparum
malaria is prevalent. Cases of serious cardiotoxicity have been reported.
It inhibits erythrocytic stages of chloroquine-sensitive and
chloroquine-resistant P. falciparum and other Plasmodium spp.
in vitro at concentrations of 0.4–4.0 mg/L. It is more active
than mefloquine and the combination of proguanil and atovaquone
against P. falciparum, but less effective than mefloquine
or chloroquine against P. vivax.
Resistance in P. falciparum has been reported in Central and
West Africa, where it has been used widely. Cross-resistance
with mefloquine has been reported in Thailand, where it has
not been used.
Structurally, halofantrine differs from allother antimalarial drugs. It is a good example of drug designthat incorporates bioisosteric principles as evidenced by thetrifluromethyl moiety. Halofantrine is a schizonticide and has no affect on the sporozoite, gametocyte,or hepatic stages. Both the parent compound and Ndesbutylmetabolite are equally active in vitro. Halfantrine’sspecific mechanism of action against the parasite is notknown. There is contradictory evidence that its mechanismranges from requiring heme to disrupting the mitochondria.There is a prominent warning that halfantrine can affectnerve conduction in cardiac tissue.
A phenanthrene methanol, formulated as the hydrochloride
for oral administration. Parenteral formulations are not
available. The enantiomers have equivalent activity in vitro.
Aqueous solubility is extremely low.
Absorption shows high intra- and inter-subject variability
and depends on co-administration with fats. Bioavailability is
increased more than six-fold after a fatty meal or by lipidbased
formulations. Bioavailability is significantly lower in
patients with malaria than in healthy individuals. Peak plasma
levels are variable and occur 3–6 h after administration.
Unlike many other antimalarials, halofantrine is not concentrated
by infected or uninfected erythrocytes. Distribution to
lipoproteins is stereo-selective. About 20–30% of the dose is
metabolized to an N-desbutyl derivative by cytochrome P450
(CYP) 3A4 and 3A5. The elimination half-life of the parent
drug is generally 1–2 days and that of the metabolite 3 days.
Little unchanged drug is excreted in urine.
Treatment of multidrug-resistant falciparum malaria
Its use has been questioned due to the existence of safer alternatives.
Its use has been questioned due to the existence of safer alternatives.
Abdominal pain, diarrhea and pruritus are the most frequent.
High doses (24 mg/kg) induce prolongation of the PR and
QTc intervals; this is not stereo-selective. There are individual
reports of fatal cardiac arrest and torsade de pointes.
To reduce the risk of cardiac toxicity it should be taken on
an empty stomach. It should not be administered with other
antimalarials that have the potency to induce cardiac arrhythmias
(mefloquine, chloroquine, quinine). Halofantrine has
also been associated with intravascular hemolysis.
Halofantrine is considered to be an alternative drug for treatment of both chloroquine-sensitive
and chloroquine-resistant Plasmodium falci parum malaria, but its efficacy in mefloquine-resistant
malaria may be questionable. The drug is metabolized via N-dealkylation to desbutylhalofantrine
by CYP3A4. The metabolite appears to be several-fold more active than the
administered drug.
Preparation Products And Raw materials
Raw materials
HALOFANTRINE manufacturers


Related Product Information
- halofantrine hydrochloride
- 2-Chloronaphthalene
- 9-ETHYLPHENANTHRENE
- 1,6-DIMETHYLNAPHTHALENE
- HALOFANTRINE
- 1-(2-NAPHTHYL)ETHANOL
- 3-(DIMETHYLAMINO)-1-(NAPHTHALEN-2-YL)PROPAN-1-OL
- 3-(NAPHTHALEN-2-YL)PROPAN-1-AMINE
- 3-(NAPHTHALEN-1-YL)PROPAN-1-AMINE
- Halofantrine HCL
1of4
PROMPT×
PROMPT
The What'sApp is temporarily not supported in mainland China
The What'sApp is temporarily not supported in mainland China
Cancel
Determine